Can a Disulfide Bond Oxidize Again
Abstract
Disulfide bonds play a key role in stabilizing protein structures, with disruption strongly associated with loss of protein function and activity. Previous data have suggested that disulfides show only modest reactivity with oxidants. In the current study, we report kinetic data indicating that selected disulfides react extremely rapidly, with a variation of 104 in rate constants. Five-membered ring disulfides are particularly reactive compared with acyclic (linear) disulfides or six-membered rings. Particular disulfides in proteins also show enhanced reactivity. This variation occurs with multiple oxidants and is shown to arise from favorable electrostatic stabilization of the incipient positive charge on the sulfur reaction center by remote groups, or by the neighboring sulfur for conformations in which the orbitals are suitably aligned. Controlling these factors should allow the design of efficient scavengers and high-stability proteins. These data are consistent with selective oxidative damage to particular disulfides, including those in some proteins.
Introduction
Disulfide bonds play a critical stabilizing role in many protein structures by forming cross-links between different regions of polypeptide chains. 21% of the ~90,000 protein structures of the Protein Data Bank contain at least one disulfide bridge, and this incidence is considerably higher in many structural and extracellular matrix proteins and receptors. Furthermore, cysteines involved in disulfide bonds are the most evolutionarily-conserved amino acids (unlike free cysteine), and in 99% of cases these are only replaced as pairs1. Disulfides serve as (largely permanent) molecular "staples" that direct and stabilize the three-dimensional structure of proteins, and determine the distance and angle constraints between the joined cysteine residues, therefore stabilizing the folded state with respect to the unfolded form2. Disulfides can also play a functional and transient role in enzyme activity3,4, such as in redox-active proteins that undergo thiol-disulfide interconversion (e.g., the thioredoxin superfamily). In these cases, the disulfide formed on oxidation of vicinal thiols, is often found in (low abundance) high-energy strained conformations (e.g., the so-called +/− RH Hook and –RH Staple configurations), which are believed to facilitate rapid reduction back to the di-thiol form5,6. Such redox-active disulfides have been shown to be important for normal cellular function, with perturbations postulated to be involved in pathological conditions that are characterized by abnormal/altered redox states, including ageing, cardiovascular disease, some cancers, asthma, rheumatoid arthritis, cystic fibrosis and multiple neurodegenerative diseases (e.g., Alzheimer's, Parkinson's, Huntington's, Creutzfeldt-Jakob disease)7.
Chronic inflammation is a well-established contributor to tissue damage associated with disease8. Chronic inflammation can arise from excessive or continued stimulation of immune cells from ongoing infection and/or incomplete removal of stimulants (e.g., particulate matter, oxidized materials)9, with this resulting in ongoing damage to tissues, with subsequent poor repair and remodelling (reviewed)8. Stimulation of neutrophils, monocytes and macrophages results in the assembly and activation of complexes that generate oxidants, including NADPH oxidases (NOXs) that generates superoxide radicals (O2 −·) and hence H2O2 by dismutation, and nitric oxide synthases that produce nitric oxide (NO·)10,11. Reaction of O2 −· with NO· generates the powerful oxidant peroxynitrous acid (ONOOH)12. Stimulated neutrophils, monocytes and some tissue macrophages release myeloperoxidase that utilises H2O2 to generate powerful two-electron oxidants [e.g. hypochlorous acid (HOCl), hypobromous acid (HOBr), hypothiocyanous acid (HOSCN)] and radicals13,14. These processes are critical to efficient pathogen killing, but result in significant collateral damage to host tissues15. Oxidant formation can also arise via electron-leakage from mitochondria, from enzymes such as xanthine oxidase and lipoxygenases, and by uncoupling of enzymes such as endothelial and inducible nitric oxide synthases (NOS) (reviewed)8. Exogenous factors can also generate oxidants, including radiation and sunlight exposure (via singlet oxygen, 1O2, formation), metal ions, and environmental pollutants (e.g., chemicals, drugs, solvents, particulates, cigarette smoke). Consequently, chronic inflammation and increased oxidant levels have been linked with multiple human pathologies8.
Radical-mediated damage to biological targets has been studied extensively, but less is known about the reactions of HOCl, HOBr, HOSCN, ONOOH and 1O2 12,14,15,16. Proteins are major targets for these oxidants as a result of their abundance, and high rate constants for reaction, with sulfur-containing amino acids being particularly prone to modification due to the presence of the reactive sulfur center (reviewed)15,17,18. Oxidation of cysteine (Cys) and methionine (Met) residues is relatively well understood (reviewed)19,20, but modification of disulfides (e.g. cystine), and the factors that control this, are less well characterized, despite the critical importance of such bonds in maintaining protein structures. Disulfide bond modification is postulated to be a key factor in determining the shelf life and activity of protein- and peptide-based medicines including antibodies and vaccines21,22, in food processing and spoilage23, in amyloid and aggregate formation24,25 and in some human diseases26,27.
In the present study we show that the apparent second-order rate constants (k 2 ) for oxidation of disulfide bonds (both low molecular mass and also in proteins) vary by up to 104-fold, and with multiple oxidants, with the variations arising from interactions between the reaction center and nearby heteroatoms. Theoretical computations have been used to help rationalize the experimental observations.
Results
Apparent second-order rate constants for reaction of HOCl with linear (acyclic) disulfides
Rate constants (k 2 ) for reactions of HOCl with acyclic disulfides were determined under pseudo first-order conditions, with the disulfide in excess, using direct stopped-flow with UV detection (230–320 nm). The resulting data were analyzed by either plotting k obs values determined from a single exponential fit (Pro-Data Viewer) to single wavelength data (between 230–310 nm) against the disulfide concentration, or by global analysis over the complete spectral range to directly determine k 2 . Kinetic data were obtained at pH 7.4 and 10 °C, and for a limited number of the disulfide compounds also at 22 °C, with the lower temperature used to slow the reactions and enhance the accuracy of the data.
k 2 for reaction of HOCl with the acylic disulfide 3,3′-dithiodipropionic acid (DTDPA) was determined as (1.7 ± 0.1) × 105 M−1s−1, consistent with previous results [(1.8 ± 0.2) × 105 M−1s−1 at 22 °C and pH 7.4]28. At 10 °C, quenching plots gave k 2 (1.04 ± 0.09) × 105M−1s−1, and global analysis (9.0 ± 1.7) × 104M−1s−1. Analogous experiments with other acyclic disulfides including bis-(2-hydroxyethyl) disulfide (2-OH-SS), cystine, cystamine, (N-Ac-Cys)2, (Cys-Gly)2, (Gly-Cys)2 and GSSG (Fig. 1) gave k 2 values between 6 × 103M−1s−1 [for (N-Ac-Cys)2] and 4.5 × 104M−1s−1 (for (N-Ac)2-GSSG) at 22 °C. The values at 10 °C were lower as expected. All the resulting rate constants are presented in Fig. 2. k 2 for reaction of HOCl with (N-Ac)2-GSSG (due to limited availability) and 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB; where the absorbance bands of HOCl and DTNB overlap) were determined using competition kinetics with Fmoc-Met (methionine tagged with the fluorescent Fmoc group on the N-terminus) as the competitive substrate, with the yields of the parent (Fmoc-Met) and its oxidation product, Fmoc-Met sulfoxide (Fmoc-MetSO) determined by ultra performance liquid chromatography (UPLC) analysis28.

Kinetic data obtained for the reaction of disulfides with HOCl.
(a) Decrease in absorbance at 270 nm (solid line) detected on reaction of bis-(2-hydroxyethyl) disulfide (2-OH-SS; 1 mM) with HOCl (0.5 mM) in phosphate buffer (0.1 M, pH 7.4) at 22 °C, with the corresponding exponential fit (dotted line). (b) Time dependent spectral changes for the reactions indicated in (a) over the spectral range 230–350 nm at 10 nm intervals over 0.5 s. (c) Pseudo-first order plots of k obs versus substrate concentration for reaction of disulfides and HOCl in phosphate buffer (0.1 M, pH 7.4, 10 °C). Data for trans-4,5-dihydroxy-1,2-dithiane (4,5-(OH)2-dithiane, ▲), Gly-Cys (▼) and GSSG (◆) were determined with 50 μM HOCl, whilst those for cystamine (○) and 2-OH-SS (●) used 0.5 mM HOCl to improve signal intensity. Each data point is k obs from an individual sample, with the error bars representing the standard deviation from n > 3 fresh aliquots.

Apparent second order rate constants for reaction of oxidants with disulfide bonds.
Apparent second-order rate constants for reaction of HOCl with cyclic disulfides
Rate constants for cyclic disulfides were determined using competition kinetics due to the rapidity of these reactions (e.g. Figs 2 and 3a,b). k 2 for the 6-membered ring compound trans-4,5-dihydroxy-1,2-dithiolane was determined as (5.9 ± 0.2) × 104 M−1s−1 (10 °C, pH 7.4), in line with the data for many of the acyclic disulfides (Fig. 2). In contrast, the 5-membered ring compounds α-lipoic acid, α-lipoamide, and 4-methyl-1,2-dithiolane-4-carboxamide (4-Me-dithiolane-CONH2) yielded k 2 values of (1.5 ± 0.2) × 108, (9.6 ± 1.3) × 107 and (7.7 ± 1.1) × 106 M−1s−1, respectively, with these being up to 20,000-fold higher than for the acyclic disulfides (Figs 2 and 3).

Competitive kinetic analysis of the reactions of (a,b) HOCl (2 μM) with α-lipoic acid and Fmoc-Met (5 μM) and (c,d) HOCl (0.2 μM) with insulin and Fmoc-Met (0.5 μM). Conversion of Fmoc-Met to Fmoc-Met sulfoxide (Fmoc-MetSO; peaks shown in a,c) was monitored by UPLC, with data from a single representative experiment shown. Plots (b,d) show data (mean ± SEM) from at least six replicates.
Apparent second-order rate constants for reaction of HOCl with proteins
Rate constants were determined for reaction of HOCl with insulin and α-lactalbumin, which contain multiple disulfide bonds but do not contain any free Cys residues that would be a competitive target for this oxidant. The values of k 2 determined (at 22 °C, pH 7.4) using competition kinetics were (7.9 ± 1.1) × 105 M−1s−1 for insulin (Figs 2 and 3c,d) and (2.5 ± 0.6) × 107 M−1s−1 for α-lactalbumin (Figs 2 and 4).

Plots derived from competitive kinetic analysis of the reactions of HOCl (2 μM) with α-lipoic acid (○), α-lipoamide (▲), lactalbumin (◼), 4-Me-dithiolane-CONH2 (⚫) or DTNB (□) in the presence of Fmoc-Met (5 μM).
Data are mean ± SEM from six replicates (where error bars cannot be seen they are smaller than the symbol). Inset shows data on an expanded x-axis for α-lipoic acid ( ○ ), α-lipoamide (▲), lactalbumin ( ◼ ), showing linearity even at low scavenger concentrations.
Apparent second-order rate constants for reaction of HOBr and HOSCN with disulfides
In the light of the variation in k 2 values for HOCl, similar experiments were carried out with HOBr and HOSCN and a limited number of disulfides, using either direct stopped-flow experiments or competition kinetics. As with HOCl, a marked enhancement in k 2 was observed with the 5-membered cyclic disulfides, α-lipoic acid and α-lipoamide with HOBr (Fig. 2), whereas with the less reactive oxidant HOSCN, no significant enhancement was detected. High k 2 values were also determined for reaction of HOBr with α-lactalbumin, and for HOSCN with both proteins (Fig. 2).
Computational modeling of reactions
The variation in k 2 for the acylic disulfides (~102), and between the acyclic species/6-membered ring structures and the 5-membered rings (~104), prompted investigation of the possible reasons for this variation. Calculation of the thermodynamics of reaction of a range of disulfide structures with HOCl, assuming the mechanism illustrated in equation 1, was carried out using the MO6-2X method and the SMD continuum model for solvent water.
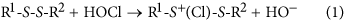
The resulting calculated reaction endothermicities at 298 K (Fig. 5) show considerable variation with both the nature and ionization state of the included functional groups (carboxylic acid/anion, neutral and protonated amine, and hydroxyl, chosen to model some of the compounds included in Fig. 2), and whether R1 and R2 were linked in a 5- or 6-membered ring. The 5-membered ring structure had the lowest calculated endothermicity (11.5 kJ mol−1), followed by the 6-membered ring (17.1 kJ mol−1). For the acyclic species, the neutral diamine (neutral cystamine) had the lowest value (28.4 kJ mol−1), with this increasing markedly with both single (49.7 kJ mol−1) and double (77.7 kJ mol−1) protonation, consistent with expectations based on an increasingly unfavorable interaction with the positively-charged sulfur center. Variation in the calculated values also occurs with the dicarboxylic acid species, 3,3′-dithiodipropionic acid (DTDPA), with the lowest values arising from the di-anion, with these increasing on mono- and di-protonation to the neutral carboxylic acid. The variation in this case is much less marked than with the amine substituents (~17 kJ mol−1 for the anion/acid, compared with ~50 kJ mol−1 for the amine series). The endothermicity calculated for the dicarboxylate is considerably lower than for the di-hydroxyl species (2-OH-SS) (32.9 versus 47.9 kJ mol−1), but comparable to the calculated value for dimethyldisulfide (31.6 kJ mol−1), which is neutral and does not include the strongly electron-withdrawing OH groups. These data are also consistent with interaction with the positively-charged sulfur center being increasingly favorable in going from neutral to monoanionic to dianionic substituents.

Calculated reaction enthalpies in kJ mol−1 for the reaction: R1-SS-R2(aq) + HOCl(aq) → R1-S+(Cl)S-R2 (aq) + HO−(aq).
The role of ring conformation in modulating the reaction kinetics was investigated by initially examining the CH3S–+S(Cl)CH3 system as a model for the chlorine adduct. In particular, quantum chemistry computations at the M05-2X/6-31G(d) level in conjunction with the solvation model based on density (SMD) continuum solvation model, were used to calculate geometries as a function of the CS–SCl dihedral angle. The variation in vibrationless energy as a function of the CS–SCl dihedral angle was then obtained through single-point calculations at these geometries at the M06-2X/6-311 + G(3df,2p) + SMD solvation (M05-2X/6-31G(d), water) level. The data presented in Fig. 6 indicate that the preferred conformations correspond to CS–SCl dihedral angles of between 60–90° and approximately 270°. The fully optimized structures have dihedral angles of 69° and 273°, respectively, with the latter lying lower in energy by 4.6 kJ mol−1. In such conformations, the p-type lone pair on sulfur is approximately parallel to the S–Cl bond. Such conformations facilitate stabilizing electron donation from the sulfur lone pair into the antibonding S–Cl orbital (Fig. 7), analogous to the interaction responsible for the anomeric effect in carbohydrate chemistry29,30,31.

Conformational energies, aNBO interaction energies, band reaction energiesc (kJ mol−1) calculated as a function of the CS–SCl dihedral angle in MeSS(Cl)Me+.

Stabilization by electron donation from the sulfur lone pair into the antibonding S–Cl orbital.
In order to quantify such interactions, the natural bond orbital (NBO) method32 has been used to determine interaction energies between a sulfur lone pair and the antibonding orbital of the adjacent S–Cl bond (Figs 6 and 8). It can be seen that the best NBO interaction energies correspond to CS–SCl dihedral angles of 90° and 270°, in line with the best optimized structures. More generally, the NBO interaction energies correlate well with the calculated conformational energies (Fig. 6). Figure 6 also contains the calculated reaction energies for equation 2, with these data indicating that there is a good correlation between the calculated reaction energies and the calculated NBO interaction energies.

Schematic representation of the interaction between natural bond orbitals corresponding to (a) the sulfur lone pair and (b) the antibonding orbital of the adjacent S–Cl bond (M05-2X/6-31 G (d) with SMD continuum solvation), and optimized structures of the chlorine adducts for (c) the 5-membered ring and (d) the 6-membered ring (M05-2X/6-31 G (d) with SMD continuum solvation).
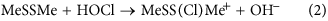
These data provide important insights into the behavior of the 5- and 6-membered ring systems. We start by noting that the calculated CS–SCl dihedral angles are 89° in the 5-membered ring adduct and 102° in the 6-membered ring adduct (Fig. 8c,d). The computed NBO interaction energies are 100.5 and 83.7 kJ mol−1, respectively. These results are consistent with expectations based on the results for the model system: the better alignment of the orbitals in the 5-membered ring adduct (dihedral angle very close to the optimum 90°) results in a more favorable interaction energy than in the 6-membered ring adduct (dihedral angle of 102°).
The reactivity of the 5- and 6-membered ring systems with HOCl can therefore be rationalized in terms of the preferred conformations. Specifically, the structural constraints in the cyclic systems lead to more effective electronic interaction between the sulfur lone-pair and the antibonding orbital of the adjacent S–Cl bond in the chlorine adduct of the 5-membered ring than in the 6-membered ring.
Discussion
Sulfur-centered amino acids, both free and in peptides/proteins, are major targets for one-electron (radical, e.g., HO ⋅ , RO ⋅ , ROO ⋅ ) and two-electron (e.g., H2O2, ONOOH, HOCl, HOBr, HOSCN, 1O2, O3) oxidants20,33. Multiple studies have shown that Cys and Met residues (both free, and in proteins) are rapidly oxidized, and are major targets20,33,34. In contrast relatively little is known about the oxidation of cystine (free or in proteins), with most studies focused on -S-S- bond reduction. The limited data available indicate that thiosulfinates (R-S-S(=O)-R'; disulfide S-oxides) are the major initial oxidation products35,36,37,38, but that these react further, resulting in -S-S- bond cleavage and sulfonic acid (RSO3H) formation. Thiosulfinates have been detected on some low-molecular-mass disulfides and proteins35,36,37,38. These are thought to arise via initial adduct (-S-S+(X)-) formation and subsequent rapid hydrolysis (equation 3, X = Cl, Br, SCN), with the latter reaction being rapid (due to the high concentration of water in most biological systems) and irreversible.
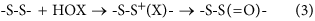
The biological significance of disulfide oxidation has been previously unclear, due to the paucity of kinetic data for these reactions. We have reported previously that acyclic disulfides react with HOCl and HOBr with moderate k 2 values (1–3 × 105 M−1s−1;39,40), but the effects of structure and electronic factors had not been elucidated, though it has been noted that ONOOH reacts with α-lipoic acid more rapidly than with GSSG and cysteine41,42. Considerable variation has, however, been reported for disulfide reactions with 1O2 43. The reactivity of Cys with oxidants is known to be pH and environment sensitive, with this arising partly from the increased reactivity of the thiolate (RS−) over the neutral form33, but also from electronic and structural factors [cf. k 2 for reaction of H2O2 with the active site Cys of peroxiredoxins of ~107 M−1 s−1, compared with ~3 M−1 s−1 for the Cys-34 residue of BSA]33,44.
The current study shows that disulfides also show large variations in k 2 , but for alternative reasons. For acyclic species, this variation is ~2 orders of magnitude, with this increasing to >4 orders of magnitude when the acyclic species are compared with the 5-membered ring disulfides. Of particular note is the increased reactivity associated with stabilization by appropriately positioned heteroatoms, and the much higher values for 5-membered ring species compared with 6-membered rings and acyclic species. For the acyclic species, this increased reactivity arises from interactions of suitably positioned electron-donor groups with the incipient positive charge developed at the sulfur reaction center, resulting in stabilization of this species (cf. Fig. 7). Enhanced reactivity is also seen with HOBr, and to a lesser extent HOSCN with proteins. The smaller effects seen with HOSCN at this pH (7.4), may be associated with differences between the attacking species, with this being the neutral species for HOCl and HOBr (pKa 7.6 and 8.7, respectively ref. 14), and − OSCN for HOSCN (pKa 4.85)45. However, even with − OSCN, a significant enhancement is apparent, as k 2 values for other disulfides are too low to be measured accurately (i.e., ≪103 M−1 s−1)46. These data, together with the computed reaction endothermicities, indicate significant and complex pH dependencies for these reactions arising from the protonation/deprotonation equilibria of both the oxidant and the target disulfides (Fig. 5). Thus the local environment of a disulfide markedly affects its reactivity.
Disulfide bond conformation also has a dramatic effect and drives the large increases in k 2 between 5- versus 6-membered rings; these differences are consistent with the calculated endothermicities. This enhanced reactivity is rationalized in terms of a highly-favorable conformation of the reactant that maximizes overlap of the p-orbitals of the sulfur atom with those of the appropriate +S-X antibonding orbital at the adjacent sulfur reaction center allowing considerable hyperconjugative stabilization. The corresponding orbitals in the 6-membered ring do not allow this stabilization as effectively. These data are consistent with the proposal47,48 that α-lipoic acid and α-lipoamide are significant targets for oxidants, and protective agents against damage to proteins with which they are associated49. Whether the resulting thiosulfinates can be repaired directly is not established, though these can undergo further reaction with GSH to give mixed disulfides and sulfenic acids, which can be repaired50 (equations 4ȓ6, Fig. 9). Disulfide oxidation to a thiosulfinate, and subsequent reaction with GSH, may therefore act as a catalytic oxidant-scavenging pathway. This pathway also provides a route to protein glutathionylation, a process strongly implicated in cellular signaling events51,52, for proteins that lack free Cys residues.

Equations 4–6.
Comparison of the kinetic data obtained for the low-molecular-mass disulfides and proteins, indicates that protein disulfides can also show increased reactivity. Both the proteins studied contain multiple disulfides. For insulin, there are two inter-chain disulfides (A7-B7 and A20-B19) between the A and B chains, and one intra-chain linkage (A6-A11). Each is important for the receptor binding activity of this protein, with A20-B19 the most important, and A6-A11 bond the least53. The value of k 2 determined for reaction of HOCl with insulin is significantly larger than that determined for (N-Ac-Cys)2 and 2-OH-SS, slightly faster than others (such as DTDPA and GSSG), and comparable with that for cystamine. These data suggest that protein-dependent factors enhance the reactivity of one (or more) of the disulfides in the protein, though the current data do not allow identification of which. A similar increased reactivity was seen with HOSCN (k 2 7.7 × 105 M−1s−1 for insulin, compared with ~103 M−1s−1 for the model compounds), but not with HOBr.
Bovine α-lactalbumin has four disulfide bonds that are important in protein aggregation and thiol-disulfide exchange reactions54. The k 2 values for reaction of this protein with HOCl, HOBr and HOSCN are markedly higher than for the acyclic disulfides. Although α-lactalbumin contains Met residues, which may contribute to the increased reactivity with HOCl and HOBr, this cannot account for the HOSCN data, as this oxidant does not react significantly with Met46. The increases in k 2 are consistent with one (or more) of the α-lactalbumin disulfides being present in a conformation that favors orbital overlap and stabilization of the incipient charged sulfur center, in a manner similar to the 5-membered ring compounds.
The conclusion that favorable conformations result in rapid reaction is consistent with data indicating that some protein disulfides exist in strained conformations5,6. These structures are particularly abundant in proteins with catalytic disulfides (e.g. thioredoxins) or those that have allosteric actions6,55. These previous studies have focused on disulfide reduction, rather than oxidation as examined here, but it is clear that there are factors that enhance both reactions. These may differ and warrant further study. Irrespective of this, it is clear that the reactivity of protein disulfides is dramatically affected by local structure and conformation. As disulfides are critical to maintaining protein structure, the current data indicate that some proteins, and sites within proteins, are particularly prone to oxidative modification and loss of function. Further studies are clearly warranted to extend these observations to a wider range of proteins, and also to proteins present in both folded and unfolded states.
Disulfide oxidation may be of particular relevance for extracellular proteins, as these typically have few Cys, large numbers of cystines, and modest numbers of Met residues. This is exemplified by human serum albumin, which has a single Cys, 6 Met and 17 disulfides (cf. UniProt entry P02768), and many extracellular matrix proteins (e.g., laminins, fibrillins and fibulins). Many extracellular matrix protein disulfides have critical functional roles, particularly with regard to the formation of receptor structures and epidermal growth factor (EGF) domains. Laminins are major components of most basement membranes, and are critical determinants of tissue architecture and cell binding, and contain up to 12% cystine residues. Fibrillins have even higher abundances, with fibrillin-1 containing 47 EGF–like domains each containing three conserved cystine bridges, with mutations in these cystine bridges associated with Marfan syndrome, the most common disorder of connective tissues56,57. Fibulins are also exceptionally cystine-rich, with this protein containing both large numbers of similar EGF domains, and an Arg-Gly-Asp (RGD) sequence critical for cell adhesion through interactions with integrins. As such, it has been strongly associated with cardiovascular disease58 and protection against cancer cell metastasis59.
Conclusions
Overall, the data presented here suggest that oxidant-mediated damage to disulfides in proteins can be particularly rapid and may play a critical role in tissue dysfunction. This is likely to be particularly true for extracellular proteins, or domains, which are cystine-rich and can be exposed to high levels of oxidants, as a result of the low levels of extracellular defense and repair systems, and the long half-life and slow turnover of extracellular matrix proteins and proteoglycans.
Materials and Methods
Reagents
(±) α-Lipoic acid, 3,3′-dithiodipropionic acid (DTDPA), bis-(2-hydroxyethyl) disulfide (2-OH-SS), L-glutathione disulfide (GSSG), cystamine dihydrochloride, trans-4,5-dihydroxy-1,2-dithiane (4,5-(OH)2-dithiane), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), α-lactalbumin and recombinant human insulin (from Saccharomyces cerevisiae) were obtained from Sigma-Aldrich (Castle Hill, NSW, Australia). Fmoc-Met, Fmoc-MetSO, and the disulfides (N-Ac-Cys-OH)2, (H-Cys-Gly-OH)2, (H-Gly-Cys-OH)2, (Boc-Cys)2 were from Bachem (Bubendorf, Switzerland). (±) α-Lipoamide was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). di-N-acetyl-glutathione disulfide [(N-Ac)2-GSSG] was from Auspep (Tullamarine, Vic, Australia). 4-Methyl-1,2-dithiolane-4-carboxamide (4-Me-dithiolane-CONH2) was from Ambinter (GreenPharma, Orleans, France). Samples were prepared in 0.1 M phosphate buffers (pH 7.4) using Nanopure water (four-stage Milli-Q system; Millipore, NorthRyde, Australia). Buffers were treated with washed Chelex resin (Bio-Rad, Gladesville, NSW, Australia) to remove contaminating transition metal ions.
Stopped-flow spectrophotometry
Apparent second-order rate constants (k 2 ) for reactions of oxidants with disulfides were obtained either by directly monitoring changes in absorbance using a stopped-flow system, or via competition kinetic methods using UPLC as reported previously28,60 and described in the Supplementary Information.
Competition kinetic measurements for reaction of HOCl and HOBr with disulfides using UPLC
Competition kinetic data were obtained for HOCl and HOBr using the conversion of Fmoc-Met to Fmoc-Met sulfoxide (Fmoc-MetSO) as the reference reaction as described previously28,60 and in the Supplementary Information.
k 2 for reaction of Fmoc-Met with HOBr was determined using DTDPA as the reference reaction [k 2 (1.1 ± 0.2) × 106 M−1s−1 at 22 °C]40, yielding a value at 22 °C and pH 7.4 of (6.5 ± 0.1) × 108 M−1s−1. This value was used to determine the values for the disulfides as described above for HOCl. As with HOCl, the primary oxidation products of HOBr and disulfides do not oxidize Fmoc-Met, and did not change the initial yield of Fmoc-MetSO. Details of the separation and detection methods are given in the Supplementary data.
Competition kinetic experiments for reactions of HOSCN
Rate constants for the reactions with HOSCN were determined using competition kinetics with oxidation of 5-thio-2-nitrobenzoic acid (TNB) to 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) as the reference reaction46. HOSCN (2.5 μM) was kept as the limiting reagent with a minimum 4-fold excess of disulfide and a constant concentration of TNB (10 μM). Concentrations of TNB were determined at 412 nm, using ε 14150 M−1cm−146.
Molecular modelling calculations
Standard density functional theory calculations were carried out using Gaussian 0961. Geometry and vibrational frequency calculations were carried out at the M05-2X/6-31G(d) level62 in the presence of a solvent (water) continuum computed with the SMD model63. Zero-point vibrational energies and enthalpic temperature corrections were obtained from scaled M05-2X/6-31G(d) vibrational frequencies using literature scale factors64. Single-point energy calculations were carried out with M06-2X/6-311 + G(3df,2p) + SMD solvation + thermal corrections65. Relative energies are quoted in kJ mol−1 and they represent 298 K condensed-phase enthalpies. Analysis of interactions was carried out using the natural bond orbital procedure32.
Errors and statistics
Stopped-flow data (reported with 95% confidence limits) were obtained from at least two independent experiments each using typically 5 different substrate concentrations, with 10 kinetic traces averaged at each concentration. For competition kinetic data (reported with 95% confidence limits) the experiments were repeated at least 3 times, with duplicate samples for each experiment. Prism 5.0 (v3.0; GraphPad, Inc) or Origin Pro 8.0 (Origin Lab, Northampton, MA, USA) were used for statistical analyses of the linear fits. Standard error propagation methods were used to combine the experimental uncertainties in k 2 and the slopes of the competition plots to provide the final confidence intervals for the reported k 2 values.
Additional Information
How to cite this article: Karimi, M. et al. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Sci. Rep. 6, 38572; doi: 10.1038/srep38572 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
-
Thornton, J. M. Disulphide bridges in globular proteins. J Mol Biol 151, 261–287 (1981).
-
Wedemeyer, W. J., Welker, E., Narayan, M. & Scheraga, H. A. Disulfide bonds and protein folding. Biochemistry 39, 4207–4216 (2000).
-
Azimi, I., Wong, J. W. & Hogg, P. J. Control of mature protein function by allosteric disulfide bonds. Antioxid Redox Signal 14, 113–126, doi: 10.1089/ars.2010.3620 (2011).
-
Cook, K. M. & Hogg, P. J. Post-translational control of protein function by disulfide bond cleavage. Antioxid Redox Signal 18, 1987–2015, doi: 10.1089/ars.2012.4807 (2013).
-
Hogg, P. J. Disulfide bonds as switches for protein function. Trends Biochem Sci 28, 210–214 (2003).
-
Schmidt, B., Ho, L. & Hogg, P. J. Allosteric disulfide bonds. Biochemistry 45, 7429–7433, doi: 10.1021/bi0603064 (2006).
-
Humphries, K. M., Szweda, P. A. & Szweda, L. I. Aging: a shift from redox regulation to oxidative damage. Free Radic Res 40, 1239–1243, doi: 10.1080/10715760600913184 (2006).
-
Halliwell, B. & Gutteridge, J. M. C. Free radicals in biology & medicine 5th edn, 1–905 (Oxford University Press, 2015).
-
Pierce, G. F. Inflammation in nonhealing diabetic wounds: the space-time continuum does matter. Am J Pathol 159, 399–403, doi: 10.1016/S0002-9440(10)61709-9 (2001).
-
Babior, B. M. Phagocytes and oxidative stress. Am J Med 109, 33–44 (2000).
-
Alderton, W. K., Cooper, C. E. & Knowles, R. G. Nitric oxide synthases: structure, function and inhibition. Biochem J 357, 593–615 (2001).
-
Radi, R., Peluffo, G., Alvarez, M. N., Naviliat, M. & Cayota, A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med 30, 463–488 (2001).
-
Klebanoff, S. J., Kettle, A. J., Rosen, H., Winterbourn, C. C. & Nauseef, W. M. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukocyte Biol 93, 185–198, doi: 10.1189/jlb.0712349 (2013).
-
Davies, M. J., Hawkins, C. L., Pattison, D. I. & Rees, M. D. Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid Redox Signal 10, 1199–1234, doi: 10.1089/ars.2007.1927 (2008).
-
Winterbourn, C. C., Kettle, A. J. & Hampton, M. B. Reactive oxygen species and neutrophil function. Ann Rev Biochem 85, 765–792, doi: 10.1146/annurev-biochem-060815-014442 (2016).
-
Davies, M. J. Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J Clin Biochem Nutr 48, 8–19, doi: 10.3164/jcbn.11-006FR (2011).
-
Davies, M. J. The oxidative environment and protein damage. Biochim Biophys Acta 1703, 93–109 (2005).
-
Davies, M. J. Protein oxidation and peroxidation. Biochem. J. 473, 805–825 doi: 10.1042/BJ20151227 (2016).
-
Schoneich, C. Thiyl radicals and induction of protein degradation. Free Radic Res 50, 143–149 (2016).
-
Trujillo, M., Alvarez, B. & Radi, R. One- and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radic Res 50, 150–171 (2016).
-
Torosantucci, R., Schoneich, C. & Jiskoot, W. Oxidation of therapeutic proteins and peptides: structural and biological consequences. Pharm Res 31, 541–553, doi: 10.1007/s11095-013-1199-9 (2014).
-
Jiskoot, W. et al. Protein instability and immunogenicity: roadblocks to clinical application of injectable protein delivery systems for sustained release. J Pharm Sci 101, 946–954, doi: 10.1002/jps.23018 (2012).
-
Livney, Y. D. & Dalgleish, D. G. Specificity of disulfide bond formation during thermal aggregation in solutions of beta-lactoglobulin B and kappa-casein A. J Agric Food Chem 52, 5527–5532, doi: 10.1021/jf049955h (2004).
-
Kurouski, D. & Lednev, I. K. The impact of protein disulfide bonds on the amyloid fibril morphology. Int J Biomed Nanosci Nanotechnol 2, 167–176, doi: 10.1504/IJBNN.2011.041000 (2011).
-
Takase, K., Higashi, T. & Omura, T. Aggregate formation and the structure of the aggregates of disulfide-reduced proteins. J Protein Chem 21, 427–433 (2002).
-
Chattopadhyay, M. et al. The disulfide bond, but not zinc or dimerization, controls initiation and seeded growth in amyotrophic lateral sclerosis-linked Cu,Zn superoxide dismutase (SOD1) fibrillation. J Biol Chem 290, 30624–30636, doi: 10.1074/jbc.M115.666503 (2015).
-
Solsona, C. et al. Altered thiol chemistry in human amyotrophic lateral sclerosis-linked mutants of superoxide dismutase 1. J Biol Chem 289, 26722–26732, doi: 10.1074/jbc.M114.565333 (2014).
-
Storkey, C., Davies, M. J. & Pattison, D. I. Reevaluation of the rate constants for the reaction of hypochlorous acid (HOCl) with cysteine, methionine, and peptide derivatives using a new competition kinetic approach. Free Radic Biol Med 73, 60–66, doi: 10.1016/j.freeradbiomed.2014.04.024 (2014).
-
Szarek, W. A. & Horton, D. Anomeric effect, origin and consequence. ACS Symposium Series Vol. 87 (American Chemical Society, Washington, DC, 1979).
-
Kirby, A. J. The anomeric effect and related stereoelectronic effects at oxygen (eds K. Hafner et al.) pp. 1–37 (Springer-Verlag, 1983)
-
Thatcher, G. R. J. The anomeric effect and associated stereoelectronic effects. ACS Symposium Series Vol. 539 (American Chemical Society, Washington, DC, 1993).
-
Weinhold, F. & Carpenter, J. E. The Structure of Small Molecules and Ions (eds R. Naaman & Z. Vager ) 227–236 (1988).
-
Ferrer-Sueta, G. et al. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem Res Toxicol 24, 434–450, doi: 10.1021/tx100413v (2011).
-
Schoneich, C. Kinetics of thiol reactions. Meth Enzymol 251, 45–55 (1995).
-
Giles, G. I. & Jacob, C. Reactive sulfur species: an emerging concept in oxidative stress. Biol Chem 383, 375–388 (2002).
-
Giles, G. I. et al. Reactive sulphur species: an in vitro investigation of the oxidation properties of disulphide S-oxides. Biochem J 364, 579–585 (2002).
-
Huang, K. P. & Huang, F. L. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem Pharmacol 64, 1049–1056 (2002).
-
Li, J., Huang, F. L. & Huang, K. P. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J Biol Chem 276, 3098–3105 (2001).
-
Pattison, D. I. & Davies, M. J. Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem Res Toxicol 14, 1453–1464 (2001).
-
Pattison, D. I. & Davies, M. J. A kinetic analysis of the reactions of hypobromous acid with protein components: implications for cellular damage and the use of 3-bromotyrosine as a marker of oxidative stress. Biochemistry 43, 4799–4809 (2004).
-
Alvarez, B. & Radi, R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311 (2003).
-
Trujillo, M. & Radi, R. Peroxynitrite reaction with the reduced and the oxidized forms of lipoic acid: new insights into the reaction of peroxynitrite with thiols. Arch Biochem Biophys 397, 91–98, doi: 10.1006/abbi.2001.2619 (2002).
-
Clennan, E. L., Wang, D., Clifton, C. & Chen, M. F. Geometry-dependent quenching of singlet oxygen by dialkyl disulfides. J Am Chem Soc 119, 9081–9082, doi: 10.1021/ja9720568 (1997).
-
Nagy, P. et al. Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J Biol Chem 286, 18048–18055, doi: 10.1074/jbc.M111.232355 (2011).
-
Nagy, P., Jameson, G. N. & Winterbourn, C. C. Kinetics and mechanisms of the reaction of hypothiocyanous acid with 5-thio-2-nitrobenzoic acid and reduced glutathione. Chem Res Toxicol 22, 1833–1840, doi: 10.1021/tx900249d (2009).
-
Skaff, O., Pattison, D. I. & Davies, M. J. Hypothiocyanous acid reactivity with low-molecular-mass and protein thiols: Absolute rate constants and assessment of biological relevance. Biochem J 422, 111–117, doi: 10.1042/BJ20090276 (2009).
-
Packer, L., Witt, E. H. & Tritschler, H. J. Alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med 19, 227–250 (1995).
-
Moura, F. A., de Andrade, K. Q., dos Santos, J. C. & Goulart, M. O. Lipoic Acid: its antioxidant and anti-inflammatory role and clinical applications. Curr Top Med Chem 15, 458–483 (2015).
-
Akiba, S., Matsugo, S., Packer, L. & Konishi, T. Assay of protein-bound lipoic acid in tissues by a new enzymatic method. Anal Biochem 258, 299–304, doi: 10.1006/abio.1998.2615 (1998).
-
Giles, G. I., Tasker, K. M. & Jacob, C. Oxidation of biological thiols by highly reactive disulfide-S-oxides. Gen Physiol Biophys 21, 65–72 (2002).
-
Dalle-Donne, I. et al. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid Redox Signal 10, 445–473, doi: 10.1089/ars.2007.1716 (2008).
-
Murphy, M. P. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal 16, 476–495, doi: 10.1089/ars.2011.4289 (2012).
-
Chang, S. G., Choi, K. D., Jang, S. H. & Shin, H. C. Role of disulfide bonds in the structure and activity of human insulin. Mol Cells 16, 323–330 (2003).
-
Ewbank, J. J. & Creighton, T. E. Structural characterization of the disulfide folding intermediates of bovine alpha-lactalbumin. Biochemistry 32, 3694–3707 (1993).
-
Marques, J. R., da Fonseca, R. R., Drury, B. & Melo, A. Conformational characterization of disulfide bonds: a tool for protein classification. J Theor Biol 267, 388–395, doi: 10.1016/j.jtbi.2010.09.012 (2010).
-
Schrijver, I., Liu, W., Brenn, T., Furthmayr, H. & Francke, U. Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet 65, 1007–1020, doi: 10.1086/302582 (1999).
-
Dietz, H. C., Saraiva, J. M., Pyeritz, R. E., Cutting, G. R. & Francomano, C. A. Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF-like domains. Hum Mutat 1, 366–374, doi: 10.1002/humu.1380010504 (1992).
-
Sivrikoz, E. et al. Gene Expression levels of elastin and fibulin-5 according to differences between carotid plaque regions. In Vivo 29, 229–235 (2015).
-
Tu, K. S. et al. Fibulin-5 inhibits hepatocellular carcinoma cell migration and invasion by down-regulating matrix metalloproteinase-7 expression. BMC Cancer 14, 938 (2014).
-
Skaff, O. et al. Selenium-containing amino acids are major targets for myeloperoxidase-derived hypothiocyanous acid: determination of absolute rate constants and implications for biological damage. Biochem. J. 441, 305–316, doi: 10.1042/BJ20101762 (2012).
-
Gaussian 09, Revision D01 (Gaussian Inc., Wallingford CT, 2009).
-
Zhao, Y., Schultz, N. E. & Truhlar, D. G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J Chem Theory Comput 2, 364–382 (2006).
-
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113, 6378–6396 (2009).
-
Merrick, J. P., Moran, D. & Radom, L. An evaluation of harmonic vibrational frequency scale factors. J Phys Chem A 111, 11683–11700 (2007).
-
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120, 215–241 (2008).
Acknowledgements
Financial support from the Novo Nordisk Foundation (Laureate Research Grant NNF13OC0004294 to MJD), and the Australian Research Council (through the Centres of Excellence Scheme, CE0561607, and Discovery Programs, DP140103116 and DP150101425) is gratefully acknowledged. We also thank the National Computational Infrastructure (NCI) National Facility and Intersect Australia Ltd for generous grants of computer time, EU COST Action CM1201 and the Heart Research Institute for a PhD scholarship for MK.
Author information
Authors and Affiliations
Contributions
M.K. and M.T.I. carried out experimental work and data analysis. B.C. and A.K.C. carried out theoretical computations, L.R. directed the computational studies. C.H.S. and D.I.P. performed data analysis and directed sections of the work. M.J.D. conceived the project, provided overall direction and coordination, and obtained financial support. All authors contributed to writing the paper, reviewed the data, and approved submission of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Reprints and Permissions
About this article

Cite this article
Karimi, M., Ignasiak, M., Chan, B. et al. Reactivity of disulfide bonds is markedly affected by structure and environment: implications for protein modification and stability. Sci Rep 6, 38572 (2016). https://doi.org/10.1038/srep38572
-
Received:
-
Accepted:
-
Published:
-
DOI : https://doi.org/10.1038/srep38572
Further reading
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Source: https://www.nature.com/articles/srep38572
0 Response to "Can a Disulfide Bond Oxidize Again"
ارسال یک نظر